Накопления амилоида могут быть локализованы в ткани или быть частью системного процесса. Прогрессивное накопление амилоида подрывает функции тканей и органов и проявляется клиническими осложнениями, дисфункцией тех органов, в которых он откладывается.
Содержимое
Этиология
Первичный амилоидоз (также называемый амилоидоз легких цепей иммуноглобулинов или AL-амилоидоз):
- Этиология остается неизвестной; однако все виды связаны с дискразией клональной плазматической клетки, хотя в большинстве случаев этого недостаточно, чтобы квалифицироваться как множественная миелома. Различие обычно состоит в процентной доле плазмоцитов в костном мозге. У менее 0,5% пациентов амилоидоз развивается в множественную миелому в течение болезни
- Нет основополагающих генетических, экологических или профессиональных факторов риска, которые были бы определены для первичного амилоидоза.
Не семейный вторичный амилоидоз (АА):
- Воспалительные полиартропатии составляют 60% случаев. Состояния включают ревматоидный артрит, ювенильный артрит, псориатический артрит и анкилозирующий спондилоартрит
- Хронические инфекции, такие как бронхоэктазы, подкожная инъекция запрещенных препаратов, пролежни, хронические ИМП и остеомиелит могут привести к вторичному амилоидозу
- Воспалительные заболевания кишечника, в частности болезнь Крона, могут привести к вторичному амилоидозу
- Болезнь Каслмэна является лимфопролиферативным расстройством, в котором вариант плазматической клетки может вызывать вторичный амилоидоз.
Вторичный амилоидоз (AA) (синдром семейной перемежающей лихорадки):
- Семейная средиземноморская лихорадка
- Синдромы перемежающей лихорадки, ассоциированные с рецепторами к фактору некроза опухолей
- Синдром Маккла-Уэллса
- Гипер-IgD синдром.
Вероятность развития вторичного амилоидоза в отсутствие одного из этих семейных или не семейных воспалительных заболеваний крайне мала. Риск развития его от хронических воспалительных артритов снижается в течение последних 2 десятилетий, по-видимому, из-за более эффективной терапии, которая подавляет основной хронический воспалительный артрит.
Наследственные формы амилоидоза (AF):
- Мутации транстиретина, приводящие к прогрессирующей кардиомиопатии или прогрессирующей нейропатии, или их сочетанию
- Мутация фибриногена A альфа-цепи приводит к вовлечению почек
- Мутация аполипопротеина A
- Мутация лизоцима
- Лейкоцитарный хемотаксический фактор 2 (LECT2) — амилоидное накопление, вызывающее почечную болезнь.
Необходимо проявлять осторожность, чтобы ошибочно не определить AF как AL из-за случайной моноклональной гаммопатии.
Патофизиология
1. Первичный амилоидоз (AL)
- У пациентов с AL продуцируются легкие цепи иммуноглобулина, которые от природы имеют предрасположенность к неправильному свертыванию из нативного альфа-спирального состояния в нерастворимую конфигурацию бета-складчатого листа.
- Развитие амилоидоза связано как с количеством продуцируемой легкой цепи, так и с качественной термодинамической тенденцией фрагментов легкой цепи иммуноглобулина к неправильному свертыванию в амилоидную конфигурацию.
- Значительные различия в использовании генов обнаружены в AL. У пациентов с клонами, полученными из использования гена линии 6aV лямбда VI, чаще доминируют проявления почечной недостаточности. Те, у кого клоны получены из генов лямбда 1c, 2a2 и 3r V, более склонны к сердечным и мультисистемным заболеваниям.
- Почка является основным органом-мишенью в AL. Моноклональная легкая цепь собирается и осаждается внеклеточно, что приводит к нарушению клубочковой базальной мембраны. Легкие цепи взаимодействуют с мезангиальными клетками, которые катаболизируют их на фрагменты, которые образуют амилоидные фибриллы.
- Сердечный амилоидоз напоминает идиопатическую рестриктивную кардиомиопатию, но функция длинной оси желудочка подавлена у всех пациентов с сердечным амилоидозом по сравнению с только 36% пациентов с идиопатической рестриктивной кардиомиопатией. У 2 расстройств есть отчетливые патофизиологические профили с ухудшением продольной функции, даже если заполнение левого желудочка нормальное. Амилоидоподобная инфильтрация сердца приводит также к нарушениям проводимости. Клинические признаки поражения сердца наблюдаются у 22-34% пациентов с AL. Смерть связана с сердечной причиной у более половины пациентов.
- Амилоидные отложения в vasa nervorum приводят к клиническим результатам, сходным с ишемической нейропатией, и приводят к смешанной аксональной демиелинизирующей картине. Кистевой туннельный синдром встречается у половины пациентов.
2. Вторичный амилоидоз (AA)
- Вторичный амилоидоз возникает в результате неправильной обработки сывороточного белка амилоида А (АА), который вместо того, чтобы быть разбитым на составляющие аминокислоты, не может быть разбит на фрагменты свыше 8,5 кДа, меченные амилоидным А белком. Этот белок является общим для всех форм амилоидоза, связанных с длительными инфекциями, среди которых бронхоэктаз (кистозный фиброз), остеомиелит, хронические микобактериальные инфекции, воспалительные заболевания кишечника, синдромы семейной перемежающейся лихорадки (например, семейная средиземноморская лихорадка, синдромы перемежающихся лихорадок, вызванных рецептором фактора некроза опухолей, синдром Макла–Уэльса, гипер-IgD синдром) и болезнь Кастлемана.
- Наиболее распространенными вовлеченными органами являются почки, желудочно-кишечный тракт и щитовидная железа. У пациентов, которые в течение длительного времени пережили внесемейный вторичный амилоидоз, могут развиться сердечного амилоидоза, но с частотой, значительно меньшей, чем для семейной формы и легких цепей. Наиболее распространенными поздними последствиями длительного развития амилоида АА являются диализ-зависимая почечная недостаточность.
3. Наследственный амилоидоз (AF)
- Большинство форм наследственного амилоидоза являются следствием неправильной установки унаследованной молекулы мутантного транстиретина (ТТР). Существуют и другие более редкие формы наследственного амилоидоза из-за мутаций аполипопротеина А1, аполипопротеина А2, фибриногена и лизоцима.
- Это обычно представляет собой либо семейную кардиомиопатию, либо семейную периферическую и вегетативную нейропатию.
4. Старческий амилоидоз (SSA)
- Существует форма амилоидоза, связанная с не мутировавшим (родным) TTR. Это происходит у пожилых людей и называется старческим системным амилоидозом; он ранее был известен как старческий сердечный амилоидоз. Накопление происходит исключительно в сердце.
Классификация
Типы амилоидоза
- Локализованный
- Соматическая
- Производный легких цепей иммуноглобулинов (первичный системный амилоидоз или AL)
- Производный от амилоидного белка А (AA или вторичный амилоидоз)
- Синдромы семейной перемежающейся лихорадки
- Семейная средиземноморская лихорадка
- Синдромы перемежающей лихорадки, ассоциированные с рецепторами к фактору некроза опухолей
- Синдром Маккла-Уэллса
- Гипер-IgD синдром
- Наследственные формы амилоидоза (AF)
- Амилоидоз, вызванный транстиретином (TTR)
- Нейропатия
- Кардиомиопатия
- Фибриногеновый амилоидоз
- Аполипопротеиновый амилоидоз
- Гелсолиновый амилоидоз
- Лизоцимовый амилоидоз
- Амилоидоз, вызванный транстиретином (TTR)
- Старческий системный амилоидоз (SSA)
- Диализный амилоидоз (А бета 2М).
Диагностика
У пациентов с амилоидозом часто присутствуют клинический синдром пораженного органа, такие как нефротический синдром, кардиомиопатия, вегетативная дисфункция или атипичная множественная миелома.
Важно дифференцировать первичный от вторичного и семейного амилоидоза. Только первичный амилоидоз (AL) имеет моноклональный иммуноглобулин в сыворотке или моче и ненормальное соотношение свободных легких цепей, хотя аналогичную картину можно наблюдать у пациентов со вторичным или семейным амилоидозом, у которых имеется совпадающая моноклональная гаммапатия неопределенного значения (MGUS).
Вторичный амилоидоз (АА) следует подозревать у пациента с хроническим воспалительным синдромом, таким как воспалительная полиартропатия, бронхоэктазы, остеомиелит или рецидивирующая лихорадочная болезнь (особенно, если присутствует семейная история).
Симптомы
Потеря веса, парэстезии, одышка и усталость — наиболее распространенные симптомы, связанные с амилоидозом, и являются общими для всех системных форм. Однако, эти жалобы неспецифичны.
Потеря веса более чем на 9 кг — частое явление и указывает на амилоидоз, если он связан с отеком или невропатией.
Усталость и одышка при нагрузке, которая обычно вызвана ранней сердечной недостаточностью, обычно не связаны с открытой сердечной недостаточностью и могут быть неправильно диагностированы как связанные со стрессом или функциональные. Обследование сердца на коронарный атеросклероз — нормальная практика.
Головокружение может быть следствием нефротического синдрома (гипоальбуминемия и внутрисосудистое сокращение объема) или сердечного амилоидоза (низкий сердечный выброс, несмотря на нормальную фракцию выброса на эхо). Ортостатическая гипотензия с обмороком может возникнуть, если присутствует вегетативная нейропатия.
Пациенты могут иметь хромоту челюсти, хромоту икр и конечностей, и, реже, стенокардию, если есть вовлечение коронарных артериол.
Симметричная дистальная дизестезия указывает на периферическую нейропатию и характерна для AL. У половины пациентов с периферической нейропатией будет связанный синдром кистевого туннеля, который характеризуется покалыванием, затрагивающим от первых до четвертых пальцев обеих рук.
Стеаторея типична для поражения кишечника. Может присутствовать сильное фекальное недержание, чередующееся с 3-4 днями запоров. Когда задействован верхний отдел желудочно-кишечного тракта, возникают тошнота, рвота и постпрандиальные судороги (так называемые псевдообструктивные симптомы).
Физикальное обследование
Обычно находки в ходе осмотра включают отек нижних конечностей и повышенное яремное венозное давление.
Многие результаты физикального осмотра при амилоидозе являются специфическими и диагностическими, но присутствуют только у 15% пациентов:
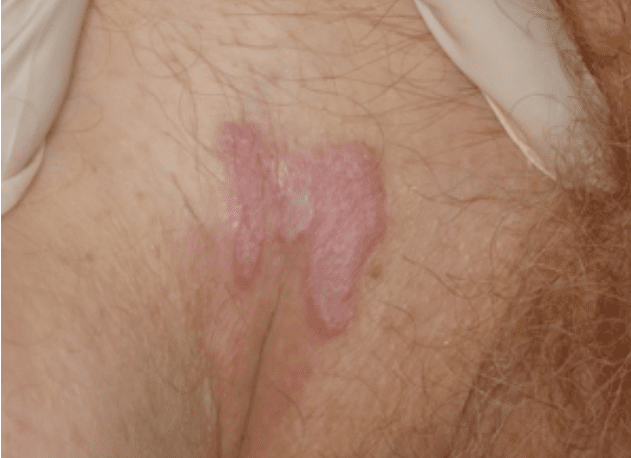
- Амилоидная пурпура присутствует примерно у 1 из 6 пациентов с АЛ. Обычно она периорбитальная, но может появляться где угодно выше линии сосков
- Петехии век распространены, но их легко упустить, если глаза пациента не закрыты
- Макроглоссия специфична для амилоидоза. Она наблюдается примерно у 10% пациентов, но ее легко упускают из виду, поскольку наиболее распространенным проявлением являются зубные отпечатки на нижней стороне языка. Макроглоссия присутствует только при AL и является диагностической
- Расширение подчелюстных слюнных желез может быть неверно истолковано как лимфаденопатия. Это типично только для AL. Причастность слюнной железы приводит к сикка-синдрому. Этим пациентам часто ошибочно диагностируется синдром Шегрена
- Пальпируемая гепатомегалия более чем на 5 см ниже правого реберного края наблюдается у 10% пациентов, а спленомегалия обычно имеет умеренную степень. Увеличение печени редко наблюдается при семейном амилоидозе, наиболее распространено при AL, и хотя печень обычно гистологически участвует в АА, ощутимая гепатомегалия является необычной для таких случаев
- Хоть встречается редко, но симптом наплечника из-за периартикулярной инфильтрации амилоидом и псевдогипертрофией специфичен для AL
- Пациенты могут проявлять диффузную мышечную слабость. Амилоидная миопатия может возникать при гипертрофии мышц вследствие внеклеточной амилоидной инфильтрации в мышце или может возникать при мышечной атрофии вследствие окклюзии сосудов, приводящей к ишемии мышц и хромоте
- Амилоидная периферическая нейропатия никогда не наблюдается при АА, наблюдается у 15% пациентов с АЛ и наблюдается у очень высокой доли пациентов с семейным амилоидозом. Симптом Тинеля (постукивая по запястному нерву на запястье, приводит к покалыванию в большом, указательном и среднем пальцах), и маневр Фалена (удерживание ладоней рук в принудительном сгибании в течение одной минуты приводит к покалыванию большого, указательного и среднего пальцев) выполняются для проверки вовлечения кистевого туннельного синдрома, если пациент жалуется на парестезию в руках.
Основные анализы для выполнения
Если клинически подозревается амилоидоз, следует провести иммунофиксацию сыворотки и мочи и анализ свободного иммуноглобулина легкой цепи, которые должны быть начальными.
Моноклональная сыворотка или белок мочи, или аномалия иммуноглобулина легкой цепи проявляются у большинства пациентов с AL и, если они положительны, нужно провести биопсию, чтобы гистологически подтвердить диагноз. Электрофорез сыворотки неадекватен, так как высокая распространенность протеинемии Bence-Jones не приводит к моноклональному всплеску при сывороточном электрофорезе.
Если результаты исследований по выявлению белка моноклонального иммуноглобулина либо иммуноглобулина свободной легкой цепи неоднозначны, необходимо провести генное тестирование для исключения семейного фибриногенового и транстиретинового (TTР) амилоидоза и тщательный осмотр для исключения причин, приводящих к возникновению вторичного амилоидоза. Прямое секвенирование амилоидных отложений методом массспектрометрии является наиболее прямым методом идентификации типа амилоидоза.
Важное значение имеет верификация амилоидных отложений с помощью биопсии. Рекомендуется использовать как подкожный жировой аспират, так и биопсию костного мозга, которые покажут накопление амилоида у 85% пациентов. Другие ткани, которые могут быть легко биопсированы, включают губы, прямую кишку, теменную артерию или кожу. Если эти исследования являются отрицательными, следует провести биопсию пораженного органа (сердца, печени, почек или нервов).
Для диагностики требуется зеленое двойное лучепреломление при окрашивании образца красным Конго. Наличие зеленого двойного лучепреломления после окрашивания красным Конго присутствует при всех формах амилоидоза, системных и локализованных. Для дифференциальной диагностике различных форм системного амилоидоза можно применять иммуногистохимические исследования амилоидных отложений. Обычно применяют коммерчески доступные антисыворотки к иммуноглобулину легких цепей, АА и ТТР, но они могут иметь как недостаточную специфичность, так и чувствительность. Во многих случаях для определения основного типа амилоида необходимо проведение масс-спектрометрии, а также иммуно-электронной микроскопии. Электронная микроскопия ограничена образцами биопсии почек для выяснения фибриллярной природы амилоида, но не используется как часть рутинной клинической практики для другого биопсийного материала. Масс-спектрометрия сейчас является золотым стандартом для типирования амилоида.
Вспомогательные исследования
Все пациенты должны иметь общий анализ крови, уровень щелочной фосфатазы в сыворотке, сывороточный альбумин, креатинин сыворотки и количественную оценку уровня белка в моче в 24-часовом образце.
Для всех пациентов требуется ЭКГ для выявления нарушений проводимости.
Изображения неспецифичны и обычно не помогают в диагностике. Однако важно оценить повреждение конечных органов, и определить прогноз. Сцинтиграфическое исследование сывороточного амилоида Р (SAP), когда оно доступно, полезно для контроля эффекта и результатов после терапии.
Эхо является необходимой частью оценки всех пациентов с доказанным амилоидозом для выявления 50% пациентов с сердечной патологией. Было доказано, что тканевой допплер и визуализация растяжения миокарда очень чувствительны для оценки дисфункции миокарда при рестриктивной кардиомиопатии.
МРТ сердца может быть полезно, если эхо неспособно дифференцировать амилоидоз от гипертрофической кардиомиопатии.
Анализы на сывороточный тропонин, натрийуретический пептид B-типа и бета-2-микроглобулин должны выполняться у всех пациентов, поскольку они обеспечивают прогностическую информацию.
Рекомендации по диагностике и обследованию, необходимые для первичного амилоидоза (AL), подчеркивают, что множественные биопсии не требуются. Оценка степени участия и дисфункции органов должна проводиться по неинвазивным критериям.
Резюме
- Амилоидоз следует подозревать у больных с:
- Недиабетическим нефротическим синдромом
- Неишемической кардиомиопатией при эхокардиограмме с гипертрофией
- Гепатомегалией или повышением щелочной фосфатазы без аномалий визуализации
- Периферической нейропатией с MGUS или CIDP
- Атипичной миеломой с моноклональными легкими цепями и умеренным плазмоцитозом костного мозга
- Пациенты, подозреваемые в наличии амилоидоза, должны подвергаться скринингу с помощью электрофореза и иммунофиксации сыворотки и мочи и анализу легких цепей без сыворотки. Если они отрицательны, врач должен узнать о возможности семейного или локализованного амилоидоза.
- Пациентам с моноклональным иммуноглобулином необходимо проводить биопсию; сначала следует брать образцы жира и костного мозга. Образцы с положительным результатом биопсии необходимо проверять методом масс-спектрометрии для подтверждения типа белка.
- Прогноз необходимо оценивать во время проведения эхокардиографии, в том числе допплерографии и деформационной эхокардиографии, а также необходимо оценивать уровни сывороточного тропонина и N-терминального фрагмента мозгового натрийуретического пептида (NT-proBNP). Всем пациентам необходимо проводить измерение иммуноглобулина свободных легких цепей.
- Пациента следует направлять на анти-плазмоцитарную терапию.
Факторы риска
- Моноклональная гаммапатия неопределенного значения
- Воспалительная полиартропатия
- Наиболее распространенная основная причина AA амилоидоза.
- Состояния включают ревматоидный артрит, ювенильный артрит, псориатический артрит и анкилозирующий спондилоартрит.
- Хронические инфекции
- Риск AA амилоидоза.
- Включает бронхоэктазы, подкожную инъекцию запрещенных препаратов, пролежни, хронические ИМП и остеомиелит
- Воспалительное заболевание кишечника
- Риск AA амилоидоза.
- В частности, болезнь Крона.
- Синдромы семейной перемежающей лихорадки
- Наиболее распространенными причинами являются семейная средиземноморская лихорадка и синдромы перемежающей лихорадки, рецептор-ассоциированные с фактором некроза опухолей, приводящие к амилоидозу АА.
- Редкие причины — синдром Маккл-Уэллса и гипер-IgD-синдром.
Дифференциальная диагностика
| Заболевание | Дифференциальные признаки/симптомы | Дифференциальные обследования |
|
|
|
|
|
|
|
| Биопсия нерва не окрашивается Конго красным . |
|
|
|
Лечение
Лечение амилоидоза зависит от его типа.
Лечение первичного амилоидоза (AL)
Целью лечения AL является подавление клона плазматических клеток, ответственного за синтез легких цепей иммуноглобулина. Прерывание отложения легких цепей позволяет организму растворять и устранять амилоидное накопление. Это предотвращает дальнейшее осаждение амилоида, которое приводит к прогрессирующей органной недостаточности.
Все пациенты с амилоидозом, подтвержденным биопсией, у которых развился висцеральный синдром (вовлечены сердце, печень, почки, нервы, легкие или кишечник), являются кандидатами на проведение терапии с трансплантацией стволовых клеток или химиотерапии. Это необходимо проводить в специализированном центре по лечению амилоидоза.
Трансплантация стволовых клеток (SCT) при AL
Пациенты, которые считаются кандидатами на SCT, обычно
- В возрасте <70 лет
- Имеют минимальную сердечную недостаточность (класс по NYHA <III) и сохраненную фракцию выброса
- Имеют креатинин плазмы ≤177 мкмоль/л (≤2 мг/дл) и
- Поражено <3 органов.
Миелоаблятивная химиотерапия с использованием мелфалана с восстановлением стволовых клеток предназначена для пациентов с низким риском осложнений, связанных с лечением. Приблизительно 20% пациентов пригодны для лечения. Однако преимущества трансплантации для амилоидоза не были полностью доказаны.
SCT — это однократная терапия. Смертность от лечения, связанная с SCT, составляет 2%. Риски включают внезапную сердечную смерть, кровотечение из желудочно-кишечного тракта и почечную недостаточность.
Рандомизированное исследование индукционной терапии с помощью бортезомиба и дексаметазона, за которым следовала SCT против изолированной SCT, выявили более высокие показатели гематологических ответов, более высокие уровни ответа и превосходную выживаемость в течение 24 месяцев.
Химиотерапия пост-SCT с мелфаланом и дексаметазоном или циклофосфамидом, дексаметазоном и талидомидом (CDT) нужна отдельным пациентам для достижения полного ответа у тех, у кого нет нормализации уровня легких цепей иммуноглобулина.
Для пациентов, которые не могут достичь гематологического ответа, исследуемые методы лечения включают использование мелфалана и дексаметазона после SCT или бортезомиба в сочетании с дексаметазоном или бортезомиба в качестве монотерапии.
Химиотерапия AL
Пациентов, которые не подходят как кандидаты на SCT, следует рассматривать как кандидатов для химиотерапии.
Комбинации химиотерапии включают мелфалан и дексаметазон, или CDT. Анализ легких цепей иммуноглобулина используется для оценки реакции на лечение и определения продолжительности терапии (обычно от 6 до 12 месяцев).
1. Пациенты с недавно установленным диагнозом
- Мелфалан и дексаметазон для пациентов, не являющихся кандидатами на SCT, эффективны в лечении долгосрочных ремиссий AL и рассматриваются как терапия первой линии.
- Комбинированная терапия CDT дает гематологический ответ 74% (21% полный, 53% частичный).
- Сообщалось также, что сочетание леналидомида с дексаметазоном является эффективным при лечении AL.
- Дексаметазон как единственный агент можно рассматривать для пациентов, считающихся слишком слабыми для исследуемой терапии, которая включает мелфалан, но показатели ответа у них ниже.
- Комбинация мелфалана, дексаметазона и леналидомида показала, что общая выживаемость в течение двух лет составила 80,8%, а выживаемость без событий составляет 53,8%. Полный гематологический ответ был достигнут у 42% пациентов, получавших дозу 15 мг леналидомида в сутки в сочетании.
- Бортезомиб обладает высокой активностью при амилоидозе с частотой от 54% до 71%, с очень быстрым средним временем ответа.
2. Отсутствие ответа
- Если есть неполный ответ на первый курс химиотерапии, возможно рассмотрение терапии бортезомибом или зачисление в клиническое испытание.
- Если предыдущее лечение с бортезомибом не удавалось, можно рассмотреть применение леналидомида и дексаметазона.
3. Рецидив
- Повторите курсы мелфалана и дексаметазона, CDT, леналидомида с дексаметазоном или бортезомиба с дексаметазоном.
Лечение вторичного амилоидоза (АА)
Лечение обычно предполагает контроль над системным воспалительным процессом. Когда амилоид обусловлен локализованной болезнью Каслмана, резекция опухоли дает эффект.
Доказано, что колхицин является эффективным в снижении частоты и продолжительности обострений заболевания (в том числе боли в брюшной полости, отека суставов, лихорадки) и предотвращает развитие АА у пациентов с семейным анамнезом средиземноморской лихорадки.
Лечение наследственного амилоидоза (AF)
Пересадка печени является наиболее признанным методом лечения наследственного транстиретинового (ТТР) амилоидоза. Среди пациентов с TTР-амилоидозом и полинейропатией после пересадки печени наблюдалась длительная регрессия в отложении амилоидов.
Лекарственными средствами, доступными для лечения пациентов с TTР-амилоидозом, являются:
- Патисиран, малая интерферирующая рибонуклеиновая кислота (siRNA), которая ингибирует продуцирование транстиретина (TTР) в печени. Вводится внутривенно в сочетании с кортикостероидами. Во время проведения 18-месячного рандомизированного контролируемого исследования было выявлено, что у пациентов с TTР-амилоидозом и полинейропатией патисиран снижает неврологический дефицит и повышает качество жизни по сравнению с плацебо.
- Инотерсен является лекарственным препаратом на основе антисмыслового олигонуклеотида, который ингибирует продуцирование TTР в печени. Вводят подкожно. Во время проведения 15-месячного рандомизированного контролируемого исследования было выявлено, что у пациентов с TTР-амилоидозом и полинейропатией инотерсен снижает неврологический дефицит и повышает качество жизни по сравнению с плацебо. В случае приема инотерсена может возникать тромбоцитопения тяжелой степени и гломерулонефрит; следовательно, перед началом лечения необходимо измерять количество тромбоцитов и почечную функцию. Поскольку есть сообщения о том, что инотерсен вызывает нарушения работы печени, перед началом лечения необходимо также измерять уровень ферментов печени. Этот препарат противопоказан: пациентам с уровнем тромбоцитов <100×10⁹/л перед началом лечения, пациентам с соотношением белок/креатинин в моче ≥113 мг/ммоль (≥1000 мг/г) или с расчетной скоростью клубочковой фильтрации <45 мл/мин/1,73 м² перед началом лечения, и пациентам с тяжелой печеночной недостаточностью. Во время лечения и в течение 8-ми недель после прекращения лечения необходимо проводить контроль количества тромбоцитов, функции почек и функциональных проб печени. В США инотерсен доступен только в рамках Программы оценки и снижения риска (REMS) в случае уверенности в преобладании пользы от применения препарата по сравнению с рисками.
- Тафамидис является стабилизатором транстиретина (TTР), который предотвращает неправильное свертывание TTР и снижает интенсивность формирования амилоидов. Во время проведения 18-месячного рандомизированного контролируемого исследования было выявлено, что у пациентов с TTР-амилоидозом и полинейропатией тафамидис замедляет прогрессирование неврологической инвалидности по сравнению с плацебо. Однако данный препарат не уменьшает количество неврологических нарушений или не улучшает качества жизни (первоначальные конечные точки). Обнаружено, что при раннем начале применения тафамидис замедляет прогрессирование неврологической инвалидности сроком до 6 лет у пациентов с TTР-амилоидозом с полинейропатией. В 30-месячном рандомизированном контролируемом исследовании относительно лечения пациентов с TTР-амилоидозом и кардиомиопатией тафамидис показал снижение смертности по всем причинам и уровня госпитализации по причине нарушений со стороны сердечно-сосудистой системы приблизительно на 30% по сравнению с плацебо. Тафамидис одобрен в Европе для лечения полинейропатии 1-й стадии у взрослых с TTР-амилоидозом
- Дифлунисал является нестероидным противовоспалительным препаратом, который показал эффективность в стабилизации TTР тетрамера и предотвращении формирования амилоидов in vitro. Во время проведения одного рандомизированного контролируемого исследования дифлунисал, применяемый в течение 2-х лет, показал снижение скорости прогрессирования неврологических нарушений и сохранение качества жизни пациентов с TTР-амилоидозом и полинейропатией по сравнению с плацебо. Дифлунисал не одобрен для применения при TTР-амилоидозе, но применяется в этих целях не по утвержденным показаниям.